




Few conditions are as harrowing as Huntington’s disease (HD). A truly devastating illness, it typically combines a triad of debilitating symptoms: a movement disorder known as chorea, which causes uncontrolled muscle spasms and loss of coordination; a decline in cognitive abilities, leading to issues such as dementia, impulsivity, and loss of focus and memory; and, in many cases, a whole host of terrifying psychiatric disturbances, including anxiety, depression, aggression, compulsive behaviour and delusions.
The disease mainly affects regions of the brain known as the striatum. This area mainly regulates motor functions, as well as other processes such as learning and inhibitory control – the ability to behave appropriately. HD usually strikes in mid-life – most often, in individuals aged 30 to 50 – and is inherited. Each pregnancy carries a 50 percent chance of transmitting the faulty gene from an affected parent, regardless of the child’s sex. It seems to occur around the world, although not all populations have been studied. The global prevalence is approximately 2.7 per 100,000 people. A high prevalence of 10.6 to 13.7 per 100,000 is observed among populations of Caucasian descent, with lower rates reported in Asian and African nations. [1]
Tragically, there is currently no cure; all that clinicians can do is try to alleviate patients’ symptoms, inevitably with diminishing returns as time goes on.

Until now, that is. Thanks to the pioneering work of Professor Edwin Chan Ho-yin and his research team from the School of Life Sciences at CUHK, a potential breakthrough in treatment of this devastating condition offers new hope to sufferers. Their research has identified a new molecular mechanism involving Poly(A) RNA polymerase D5, or PAPD5 for short, which plays a crucial role in HD. By examining the post-mortem brain tissue of HD patients, the team discovered that PAPD5 levels are at least 4.5 times higher in the brains of those patients compared to healthy individuals. They found that elevated PAPD5 causes the loss of microRNAs–tiny RNA molecules essential for gene regulation-which in turn causes neuronal cells to die.
And there’s even more good news: the team has discovered that inhibiting PAPD5 can almost completely stop this from happening. Not only that: they’ve found a highly promising potential drug candidate for the task. It’s called BCH001, which has demonstrated impressive results in cell models during preliminary testing.
“Our results demonstrate that treatment with 400 nM of BCH001 nearly completely suppresses cell death in our HD cell model, achieving approximately 90-95% reduction compared to untreated controls,” says Professor Chan.
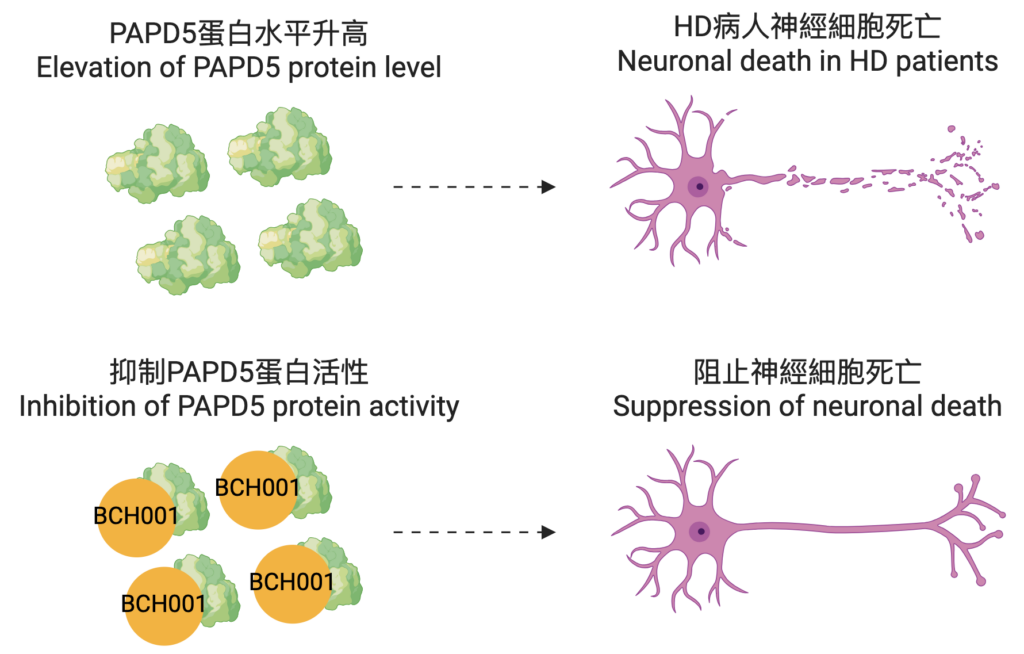
It’s been challenging to develop effective therapies in the past, he explains, because of HD’s complex nature. Although it’s caused by the mutation of a single gene, known as Huntingtin, this mutation disrupts multiple cellular pathways, complicating treatment strategies. The lack of early biomarkers means it’s difficult to detect the disease before symptoms occur; by the time symptoms manifest, significant neuronal damage has often already occurred, and it’s extremely difficult to restore lost brain function. The wide range of symptoms also means that each patient is different, complicating the design of clinical trials and the measurements of treatment effects. And, given that the disease affects the brain, many potential drugs just don’t work, because they can’t cross the blood-brain barrier. BCH001 has the potential to overcome some of these hurdles. Our discovery positions PAPD5 and its associated pathway as promising therapeutic targets for combating HD neurotoxicity, and BCH001, a small molecule inhibitor, can inhibit PAPD5 activity and suppress the pro-apoptotic pathway, demonstrating significant potential for treating this disease.
Professor Chan has been investigating Huntington’s disease for many years. “I started my research on HD in 1998,” he says. “In 2010, I decided to translate my basic research findings into therapeutic development. Over the past 15 years, my group has primarily focused on developing drugs. This current study is our first to emphasise an enzyme that is upregulated in HD patients’ brains. Identifying a small molecule drug that can target an enzyme makes this therapeutic intervention approach much more direct and specific. Our breakthroughs open up a new avenue for HD drug development.”

The team has lodged an application for a US patent to use PAPD5 and BCH001 for HD therapies. Professor Chan and his team are now in the process of screening other drug candidates to see if they can target toxic RNAs and help to treat HD.
Responses from the HD community have been very positive so far, he adds. Professor Chan has been invited to present his work at biomedical conferences in Italy and mainland China, and to share his findings through the Chinese Huntington’s Disease Association network. If everything goes as planned, he could soon have life-changing news to share with those whose lives are impacted by this devastating disease.
[1] Ajitkumar A, De Jesus O. Huntington Disease. In StatPearls. Treasure Island: StatPearls Publishing. 2024. https://www.ncbi.nlm.nih.gov/books/NBK559166/



